
Introduction
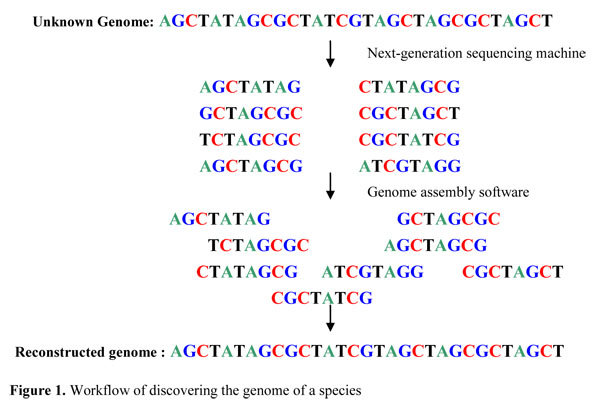
DeoxyriboNucleic Acid (DNA) is a chain of nucleotides that contains genetic information of all known living organisms. There are four kinds of nucleotides, adenine (A), cytosine (C), guanine (G) and thymine (T). Each species in the world can be represented by different DNA sequences, called genomes, with lengths vary from million (bactoria), to billion (human) and up to hundred billion (some plants). Discovering the genomes of different species is a fundamental problem of almost all molecular biology researches and discovering the genomes of every individual makes personal medicine possible. However, discovering the genome of a species, say human, is not an easy task. Over hundred of institutions have taken 13 years to discover a human genome which cost over 6 billion US dollar. Luckily, the developing of next-generation sequencing machine speeds up this process to a week with a much lower cost at around ten thousand US dollar. Given the DNA sequence of a species, the next-generation sequencing machine can generate billion of overlapping DNA fragments with length around 100 of the DNA sequence. This huge amount of data DNA fragments can be combined to reconstruct the original genome sequence using computer. And this computational problem is called genome assembly problem.
Major Difficulties
There are three major difficulties of the genome assembly problem.
(1) Repeat problem: some regions of the genome may have the same pattern. It will introduce difficulties when we determine the relative order of the DNA fragments next to these repeat regions.
(2) Gap problem: as the DNA fragments are randomly sampled from the genome, some DNA fragments may disappear. These missing fragments will prevent us from constructing the original genome.
(3) Error problem: because of experimental problem, over half of the DNA fragments produced by the next-generation sequencing machine contain errors. These errors must be corrected before reconstructing the genome.
The above problems do not appear alone; instead, they can occur on the same DNA fragment which makes the problem more complicated. For example, DNA fragments from repeat regions may contain errors such that we may notice there is a repeat region.
IDBA Algorithm
Although there are many DNA fragments with error, each error fragment contains some correct regions which can be used for reconstructing the genome. In order to obtain this information, each DNA-fragment is broken into many length-k overlapping substrings. Two length-k substrings are considered occurrence in the genome adjacently if the last k – 1 pattern of a substring is the same as the first k – 1 pattern of another substring. As those length-k substrings are usually sampled less than the correct one, IDBA can filter those substrings with error. However, what is a suitable k? Instead of finding a suitable k, IDBA sets the value of k to a small value and increases the value of k iteratively. By considering multiple values of k, IDBA can reconstruct most of the genome with higher accuracy than other assembly software.
Please visit http://i.cs.hku.hk/~alse/idba/ to learn more about IDBA.
For further information on this research topic, please contact Professor Francis Chin, Dr Henry Leung or Dr SM Yiu.